Links to external sources may no longer work as intended. The content may not represent the latest thinking in this area or the Society’s current position on the topic.
Understanding fast-ion conduction in solid electrolytes
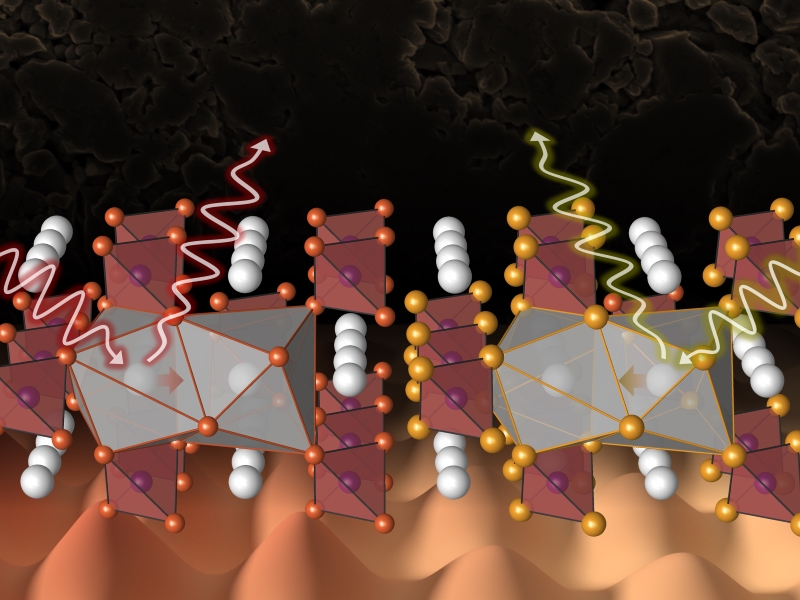
Theo Murphy meeting organised by Dr Benjamin Morgan, Dr Wolfgang Zeier and Professor Serena Corr.
'Superionic' solid electrolytes exhibit exceptional room-temperature ionic conductivities, giving them the potential to enable next-generation battery and fuel-cell technologies. Developing new high-performance electrolytes requires understanding how chemistry can control fast ionic conduction. This meeting will bring together experts on solid electrolytes, superionic conduction, and complex diffusion mechanisms in solids, to discuss the mechanistic origins of fast-ion conduction in solid electrolytes.
An accompanying journal issue has been published in Philosophical Transactions of the Royal Society A.
Attending this event
This event has taken place.
Enquiries: contact the Scientific Programmes team
Schedule
| 09:05 - 09:30 |
The role of anion dynamics in controlling ion mobility in solids
Ion mobility in solids is important for the development of solid-state batteries that employ solid electrolytes. Early studies have argued that the rotational dynamics of complex (or cluster) anions play a role in fostering cation motion, typically at temperatures above a volume-increasing polymorphic phase transformation. This talk will discuss the impact of anion dynamics on cation mobility across several ion-conducting solids. Detailed atomic-scale simulations are performed to map out the nature of anion motion and to quantify correlations with cation mobility in space and in time. Analyses are conducted to identify the circumstances under which anion rotations may occur. With this knowledge it may be possible to design solid electrolytes whose cation mobility is intentionally enhanced by anion dynamics. 
Professor Donald Siegel, University of Michigan, USA
Professor Donald Siegel, University of Michigan, USADon Siegel is Professor and Chair of the Walker Department of Mechanical Engineering at the University of Texas at Austin (UT). At UT he is a Temple Foundation Endowed Professor and holds a Cockrell Family Chair for Departmental Leadership. He is a member of the Texas Materials Institute and the Oden Institute for Computational Engineering and Sciences. Prior to joining UT in 2021, Professor Siegel spent 12 years as a faculty member at the University of Michigan. Siegel is a computational materials scientist whose research targets the development of energy storage materials and lightweight alloys. He is a recipient of the NSF Career Award and a Gilbreth Lectureship from the National Academy of Engineering. Professor Siegel received a Ph.D. in physics from the University of Illinois at Urbana-Champaign. His postdoctoral training was performed at Sandia National Laboratories and at the U.S. Naval Research Lab. In 2015-2016 he was a Visiting Professor at the Technical University of Denmark. |
|---|---|
| 09:30 - 09:45 | Discussion |
| 09:45 - 10:15 |
Atomistic mechanisms underlying non-Arrhenius superionic transport in AgCrSe2
Superionic conductors are solid-state materials that exhibit liquid-like ionic conductivities, which that makes them promising candidates for applications in energy harvesting and storage. In a subclass of these materials, the logarithm of the ion conductivity exhibits a change in its slope with respect to inverse temperature. Interestingly, these temperature-induced changes in the apparent energy barrier for ion transport occur without the materials undergoing any structural changes in their framework. In this work, the authors use classical molecular dynamics (MD) simulations to elucidate the atomistic mechanisms underlying such non-Arrhenius behaviour in the superionic conductor AgCrSe2. They first derive interatomic potential energy functions for mobile and framework ions based on trajectories collected from ab-initio MD (AIMD) simulations. Through a multistate iterative Boltzmann inversion approach, the authors develop minimal force field that reproduces the interatomic radial distribution functions, phonon density of states, and short-time Ag+ ion dynamics calculated from AIMD. The force field is then used within long timescale MD simulations to predict ionic conductivity and illuminate on the mechanism of ion diffusion. Apart from reproducing the experimentally measured conductivity of Ag+ ions in AgCrSe2, the simulations reveal how the non-Arrhenius behaviour in ion conductivity emerges from temperature-induced variations in the fraction of 'active' ions available for migration induced by an order-disorder transition in the Ag+ sublattice. The simulations also reveal the crucial role played by the repulsion between ions occupying adjacent sublattice sites in producing the order-disorder transition, intriguing temperature-dependent spatiotemporal correlations exhibited by ions, and the role of lattice flexibility in both lowering the energy barriers for ion migration and mitigating ion repulsion effects to lower the order-disorder temperature. The transport mechanisms revealed here along with the modified activated-transport model of ion diffusion proposed here should be relevant to a broad range of superionic materials. 
Professor Gaurav Arya, Duke University, USA
Professor Gaurav Arya, Duke University, USAGaurav Arya is a Professor of Mechanical Engineering and Material Science at Duke University. Prior to joining Duke in Fall 2017, he was an Assistant Professor and then Associate Professor in the Department of NanoEngineering at UC San Diego. He obtained his BTech degree in Chemical Engineering from IIT Bombay in 1998, and PhD degree, also in Chemical Engineering, from the University of Notre Dame in 2003. He carried out his postdoctoral research at Princeton University and New York University. Professor Arya’s research focuses on the development and application of molecular simulations to provide a fundamental, molecular-level understanding of various biomolecular and nanoscale systems of interest, including protein motors, chromatin, ion-transport materials, DNA nanostructures, and nanoparticle-polymer composites. |
| 10:15 - 10:30 | Discussion |
| 10:30 - 11:00 | Break |
| 11:00 - 11:30 |
From hopping to conduction: information thermodynamics of ion transport
Within information theory, heat-activated diffusion and transport can be treated as the erasure of enthalpically stabilized memory of an initial state over time. First, this conceptual framework is tested using molecular dynamics simulations of the historic model superionic conductors, the beta- and beta"-aluminas [1]. The simulated distributions of displacements approach maximum-entropy Gaussian distributions only slowly, and ion transport is sub-diffusive to macroscopic timescales. From the features and descriptors of sub-diffusion, the full production of information entropy and elimination of path-dependent effects emerge as necessary criteria for the establishment of detailed balance and ergodic behaviour. The time dependence of correlations in ion transport is tested experimentally using a non-linear optical measurement [2]. Single-cycle terahertz pulses near-resonant with phononic vibrations excite ionic hopping in solid electrolytes, and transient birefringence probes the pump-induced anisotropy of subsequent hops. The picosecond-timescale relaxation of birefringence measures the decay of orientational memory as a time-dependence of the correlation factor in ion diffusion. Finally, computational extension of the optical experiments allows for a determination of vibrational attempt frequencies for ion hopping. [1] Poletayev, Dawson, Islam, Lindenberg, Nature Materials (2022) 21, 1066-1073. 
Dr Andrey Poletayev, University of Oxford, UK
Dr Andrey Poletayev, University of Oxford, UKDr Andrey Poletayev is a postdoctoral scholar at the University of Oxford. He has completed his PhD at Stanford University. His work has focused on triggering and probing ionic conduction at picosecond timescales, and bridging atomistic to macroscopic models of ion conduction using stochastic and non-equilibrium thermodynamics. He continues to contribute to a carbon-free economy by studying energy-storage materials with atomistic simulations, electrochemistry, and ultrafast spectroscopy. |
| 11:30 - 11:45 | Discussion |
| 11:45 - 12:15 |
Probing oxide ionic diffusion by quantitative fitting of quasielastic neutron scattering data
Inelastic neutron scattering is the only experimental technique that simultaneously probes ionic diffusion (as quasielastic neutron scattering, QENS) and lattice dynamics (as a generalised density of states, GDOS). In solid-state ionic conductors (SSICs) where the diffusing species has a predominantly incoherent neutron scattering cross section (the exemplar of which is hydrogen), key parameters describing the atomistic nature of diffusion, such as jump lengths and residence times, can be extracted directly by modelling the form of the QENS. However, this is a far more challenging problem when the diffusing species have significant coherent cross-sections, such as oxygen and lithium. The author shows here that it is possible to quantitatively model coherent QENS from a SSIC. By starting with the ideal case, high-temperature cubic δ-Bi2O3, for which the diffusing species (oxygen) is an almost purely coherent scatterer, the structure is simple and the conductivity (hence the QENS signal) is high. The results show that oxide-ionic diffusion in δ-Bi2O3 is isotropic (liquid-like), even though some directions present shorter oxygen-vacancy distances, an insight corroborated by computational dynamics simulations. More broadly, they demonstrate the power of QENS for studying functional energy materials, notably for solid-oxide fuel cells and potentially lithium-ion batteries. 
Professor Christopher Ling, The University of Sydney, Australia
Professor Christopher Ling, The University of Sydney, AustraliaChris Ling is a Professor in the School of Chemistry at the University of Sydney. He has particular interests in solid-state ionic conduction at high temperatures, and strongly-correlated electronic behaviour at low temperatures. His research in inorganic solid-state chemistry makes particular use of neutron and synchrotron X-ray scattering methods to characterise the structures and dynamics of new materials. Chris is currently a Theme Leader for Functional Energy Materials in the School of Chemistry, Deputy Director of the Sydney Analytical Core Research Facility, and Chair of the IUCr Commission on Inorganic and Mineral Structures. |
| 12:15 - 12:30 | Discussion |
| 12:30 - 13:30 | Lunch |
Chair

Professor Neil Allan, University of Bristol, UK
Professor Neil Allan, University of Bristol, UK
Neil Allan has been a Professor of Physical Chemistry at the University of Bristol since 2003. He is a former Director of the Centre of Computational Chemistry (2008–2011). He has just completed five years as Chair of CCP5, the national Collaborative Computational Project for computer simulation of condensed phases. He was educated at Oxford University and then spent four years in industry working at ICI, before moving to Bristol in 1989. His main interests lie in the simulation materials of technological, geological and industrial interest, using a wide variety of theoretical techniques, ranging from atomistic lattice simulations to ab initio electronic structure calculations. Much of his work deals with the computed properties of inorganic materials and minerals, including solid solutions, the effects of additives, dopants and impurities, thin films and crystal growth.
| 13:30 - 14:00 |
Simulating Li-diffusion in amorphous and polycrystalline LLZO
The garnet Li7La3Zr2O12 (LLZO) is one of the most promising solid electrolyte candidates due to its large electrochemical stability window against a lithium anode, reasonable ionic conductivity, from 10-4 to 10-3 S cm-1, and mechanical strength. However, the effect of the polycrystalline microstructure is not completely solved. The author compares the atomic-scale diffusion mechanisms in the amorphous grain-boundaries versus the bulk grains and calculate the effect of the microstructure on the effective diffusivity. The Li-ion diffusion is simulated with molecular dynamics and the author shows that using an accurate potential is important to accurately understand the diffusion mechanism. 
Professor Nicole Adelstein, San Francisco State University, USA
Professor Nicole Adelstein, San Francisco State University, USANicole Adelstein is an Assistant Professor in the Department of Chemistry and Biochemistry at San Francisco State University. Together with her group of undergraduate and master students, she studies ionic diffusion in solid-state materials using molecular dynamics, both quantum and classical simulations. Before arriving at SFSU in 2015, Nicole was a post-doc at Lawrence Livermore National Laboratory for three years. She received her PhD from the University of California, Berkeley in Materials Science and Engineering and her BA from Reed College in Chemistry and Physics. |
|---|---|
| 14:00 - 14:15 | Discussion |
| 14:15 - 14:45 |
Lithium ion dynamics in crystalline solids: site-specific Li+ hopping in cathode materials as revealed by high-resolution 6Li 2D EXSY NMR
The visualization of atomic or ionic jump processes on the Ångström length scale is important to identify the preferred diffusion pathways in solid electrolytes for energy storage devices. 2D high-resolution 6Li NMR exchange spectroscopy (EXSY) is highly suited to yield site-specific insights into local Li+ hopping processes within a single measurement. Here, the beta-modification of Li3VF6 is used as a model system for such an investigation as it provides a range of important Li+ geometric environments in one and the same crystal structure that are useful to elucidate qualitatively a ranking of energetic preferences of the Li+ exchange processes. It turned out that Li+ ion jumps between face-shared polyhedra are preferred, followed by Li+ exchange between edge-shared configurations for which interstitial sites are needed to jump from site to site. Surprisingly, Li+ exchange between corner-shared polyhedra and Li+ hopping involving almost isolated LiF4 polyhedra do contribute to overall Li+ self-diffusion as well. In this sense, the current study experimentally verifies current predictions by theory but also extends our understanding of ion dynamics between corner-shared Li-bearing polyhedra. 
Professor Martin Wilkening, Graz University of Technology, Austria
Professor Martin Wilkening, Graz University of Technology, AustriaMartin Wilkening has been a Full Professor for Physical Solid state Chemistry at the Graz University of Technology since 2011. He is Director of the Christian Doppler Laboratory of Lithium Batteries - a collaboration between TU Graz and industrial companies such as AVL, Infineon and TDK Electronics. He studied Chemistry at the Leibniz University Hannover and obtained his doctoral degree in 2005 with a dissertation on ‘Ultraslow Li Motions in Solids’. His PhD thesis was awarded the Starck-Promotionspreis of the German Chemical Society (GDCh) and the Wissenschaftspreis Hannover. In 2009 he received the ADUC annual award of the GDCh for his contributions to spin-alignment echo NMR to directly probe the shape of single-spin motional correlation functions in solids. In his workgroup at the TU Graz, cation and anion dynamics in solids are studied with a broad range of NMR techniques and broadband conductivity spectroscopy. Specifically, his research team focusses on self-diffusion processes in nanostructured and amorphous energy materials including mechanosynthesized non-equilibrium compounds and nanoglasses. One overarching aim is to identify the origins of rapid ion exchange in model substances. His work aims to learn how the dimensionality of diffusion pathways and correlation effects influence the elementary steps of ion hopping. |
| 14:45 - 15:00 | Discussion |
| 15:00 - 15:30 | Break |
| 15:30 - 16:00 |
Combining operando techniques to probe chemo-mechanical evolution at buried solid/solid interfaces
Chemical and mechanical changes local to the electrode/electrolyte interface critically impact performance in all-solid-state batteries. Unfortunately, the dynamics at electrochemical interfaces are exceptionally challenging to probe in all-solid-state batteries because these changes take place across multiple length scales (from the nano- to meso-scale) and are buried within the system (at the solid/solid electrode/electrolyte interface). Here, the author will show their efforts to couple operando acoustic transmission measurements with nuclear magnetic resonance spectroscopy and imaging to correlate changes in interfacial mechanics with the growth of Li microstructures and the solid electrolyte interphase (SEI) in a non-invasive, multimodal fashion. Specifically, the research group studies chemo-mechanical changes at the interface between Li metal anodes and Li7La3Zr2O12 solid electrolytes as a function of stack pressure and current density. 
Professor Lauren Marbella, Assistant Professor in the Department of Chemical Engineering, Columbia University, USA
Professor Lauren Marbella, Assistant Professor in the Department of Chemical Engineering, Columbia University, USALauren Marbella is an Assistant Professor in the Department of Chemical Engineering at Columbia University. Her research group focuses on understanding the relationship between electrochemical performance and interfacial chemistry in devices for energy storage and conversion. Her research relies heavily on the use of nuclear magnetic resonance imaging (MRI) and spectroscopy to evaluate changes in material properties in real time to elucidate the chemical mechanisms underpinning degradation in Li and beyond Li ion battery systems. Marbella’s research has received numerous awards including the Cottrell Scholar Award (2022), the National Science Foundation (NSF) Faculty Early Career Development (CAREER) Award (2021), and the Scialog Collaborative Innovation Award for Advanced Energy Storage (Sloan Foundation, 2019). Marbella received her PhD in chemistry from the University of Pittsburgh in 2016, under the direction of Professor Jill Millstone. In 2017, she was named a Marie Curie Postdoctoral Fellow at the University of Cambridge in the group of Professor Clare Grey. There, she was also named the Charles and Katharine Darwin Research Fellow, which recognizes the top junior fellow at Darwin College at the University of Cambridge. She joined the chemical engineering faculty at Columbia University in 2018. |
| 16:00 - 16:15 | Discussion |
| 16:15 - 16:45 |
Understanding Li ions diffusion in sulphide- and oxide-based conductors from NMR
Significant progress is being made in the advancement of next-generation energy storage materials by implementing Li solid-state electrolytes to produce Li all-solid-state batteries. This eliminates the use of liquid electrolytes containing highly volatile and flammable organic solvents which present significant safety issues. Li-containing materials providing fast Li ion transport pathways are fundamental in Li solid-state electrolytes and the future of all-solid-state batteries. Collaborative computationally-guided materials discovery1 has provided a workflow for identifying unexplored selection of elements containing Li ions2,3 and designing Li solid-state electrolytes.
Li ions transport is the key sought physical property and, in this contribution, we will reveal several efficient NMR methods to directly probe the Li ions dynamics in a range of recently discovered sulphide2,4–6- and oxide7–9-containing materials. We exploit a range of variable temperature multinuclear (6Li and 7Li) and multidimensional NMR approaches, such as line shape analysis, exchange phenomena and relaxometry measurements, to determine the Li ion mobility pathways, including the dimensionality of the diffusion processes, and quantify Li ions jump rates. For example, these approaches deployed on Li3AlS32 identify that Li ion diffusion is fast within the tetrahedral and tetrahedral/octahedral layers but slow between these layers limiting long range translational Li ion mobility.10 These provide a framework for the further development of more highly conductive Li solid-state electrolytes such as Li4.3AlS3.3Cl0.7.5 References: (1) Collins, C. et al. Nature 2017, 280. (2) Gamon, J. et al. Chem. Mater. 2019, 9699. (3) Vasylenko, A. et al. Nat. Commun. 2021, 5561. (4) Leube, B. T. et al. Chem. Mater. 2018, 7183. (5) Gamon, J. et al. Chem. Mater. 2021, 8733. (6) Morscher, A. et al. Chem. Mater. 2021, 2206. (7) Enciso-Maldonado, L. et al. Chem. Mater. 2015, 2074. (8) Santibáñez-Mendieta, A. B. et al. Chem. Mater. 2016, 7833. (9) Han, G. et al. J. Am. Chem. Soc. 2021, 18216. (10) Duff, B. B. et al. submitted.

Professor Frédéric Blanc, University of Liverpool, UK
Professor Frédéric Blanc, University of Liverpool, UKFrédéric obtained his PhD in chemistry in 2008 from the Université de Lyon and the Centre for High Field NMR at the Ecole Normale Supérieure de Lyon, under the supervision of Professor Christophe Copéret and Professor Lyndon Emsley. He studied the structure-dynamics relationship in heterogeneous catalysis and developed new solid state NMR methods to understand the chemistry of catalysts on surfaces. He then received a Lavoisier fellowship and underwent postdoctoral work at the State University of New York in Stony Brook, NY in 2008-2010 with Professor Clare P Grey, where he got interested in the understanding of the structure elucidation of energy materials with NMR spectroscopy. He then moved to the University of Cambridge as a Marie Curie fellow in 2011 and looked at the dynamics of oxygen and protons in electrolytes by high temperature NMR spectroscopy. In December 2012, he was appointed to a lectureship in the Department of Chemistry and the Stephenson Institute for Renewable Energy at the University of Liverpool and was promoted to Senior Lecturer in 2016, Reader in 2018 and to a Personal Chair in March 2022, where he is focusing on the development and application of NMR spectroscopy in materials chemistry. Recent work has focused on enhancing the NMR signal of unreceptive nuclei such as 17O at natural abundance and on exploiting the ability of NMR to probe dynamics to investigate a range of solid state Li ion materials as Li conductors. |
| 16:45 - 17:00 | Discussion |
| 17:00 - 18:00 | Poster session |
Chair

Dr Stephen Hull, The ISIS Facility, UK
Dr Stephen Hull, The ISIS Facility, UK
Dr Stephen Hull completed his PhD in Physics at the University of Reading in 1985, followed by a Postdoctoral Researcher position held jointly between the Clarendon Laboratory, University of Oxford and the Harwell Laboratory, UK Atomic Energy Authority. In 1988, he joined the Crystallography Group of the ISIS Facility, as Instrument Scientist on the Polaris powder diffractometer. At ISIS, he established a research programme investigating structure-property relationships within ionically conducting solids, focussing on both ‘model’ superionic compounds and more technologically relevant materials for fuel cell and battery applications. In 2012 he became Head of the ISIS Crystallography Group, with responsibility for a suite of 9 diffraction and imaging instruments, which perform experiments across a diverse range of scientific research in the fields of chemistry, physics, earth science, materials science and engineering.
| 09:00 - 09:30 |
Paradigms of structural, chemical, and dynamical frustration in superionic conductors
Rationally motivated computational discovery and optimization of solid electrolytes require the development of reliable descriptors for fast solid-state ionic conductivity. However, many of the fundamental motivations for superionic behaviour in solids remain enigmatic, which has generally slowed progress in screening new candidates or tuning existing materials to maximize ionic conductivity. Dr Wood will discuss the use of high-performance computer simulations and advanced analytical techniques to unravel various mechanisms of ionic conductivity in model classes of solid electrolytes. Using computational “experiments”, the simulations systematically isolate factors such as stoichiometry, strain, composition, crystal structure, and local environment in the determination of ionic conductivity. Collectively, the results point to the importance of a frustrated energy landscape in promoting ultrafast diffusion. Different types of frustration in model superionic conductors will be discussed, arising from factors such as off-stoichiometry, competition between interstitial site occupancies, symmetry incompatibilities between local bonding character and lattice geometry, and dynamical frustration coupled to anharmonic lattice motion. Dr Wood will explore the physicochemical relevance of these factors for understanding and promoting cation mobility, with a view towards developing design rules for engineering faster ionic conductors. Among the topics to be discussed is the dependence of the different frustration paradigms on the fundamental nature of the lattice-forming ions, which suggests there may be no single universal descriptor for ionic conductivity, but rather classes of superionic conductors with similar underlying motivations. Specific examples will be drawn from recent results on superionic materials based on oxides, halides, and polyatomic anions. 
Dr Brandon Wood, Lawrence Livermore National Laboratory, USA
Dr Brandon Wood, Lawrence Livermore National Laboratory, USABrandon Wood is a Staff Scientist in the Materials Science Division at Lawrence Livermore National Laboratory (LLNL), USA. He received his PhD in Materials Science and Engineering from MIT in 2007. His primary research activities lie in the application of first-principles and mesoscale simulation techniques to materials for energy storage and conversion, including solid-state batteries, hydrogen storage media, and electrocatalysts. He is particularly interested in complex dynamics of interfaces and disordered systems. He currently serves as the Deputy Director of the LLNL Laboratory for Energy Applications of the Future (LEAF) and Theory Director of the US Department of Energy Hydrogen Materials--Advanced Research Consortium (HyMARC). |
|---|---|
| 09:30 - 09:45 | Discussion |
| 09:45 - 10:15 |
Role of local structure on ionic conductivity in bismuth oxides
Ab initio simulation techniques are increasingly able to provide often general new insights into diffusion processes in condensed phases. In this presentation links between local structure and ionic migration are explored by examining the energy landscapes, calculated ab initio, of fast-ion conductors with particular attention to bismuth oxides. A picture of the high-temperature disordered phase of δ-Bi2O3 very different from the conventional emerges from this approach. Only the mean structure is cubic; transitions between energetically accessible minima, with structures similar to low symmetry motifs in the low-temperature ordered phases are responsible for the high oxide mobility. The authors also discuss the stabilisation of thin films of δ-Bi2O3 on the (100) surface of SrTiO3. The authors then compare δ-Bi2O3 and BIMEVOX [1]. Again there are a large number of energy minima and many low-lying thermally accessible configurations. In these there are equal numbers of oxygen vacancies in each vanadium–oxygen layer, a range of vanadium coordinations and large variations in the Bi–O and V–O distances. By dividing local minima in the energy landscape into sets of configurations, the authors then examine diffusion in each different layer using ab initio molecular dynamics. These simulations show that diffusion mainly takes place in the <110> direction in the vanadium layers, involving the cooperative motion of the oxide ions between the O(2) and O(3) sites in these layers, but not O(1) in the Bi–O layers, in agreement with experiment. The calculated ionic conductivity is also in reasonable agreement with experiment. The authors also briefly contrast these Bi oxides (‘type I’ superionics) with the β-phase of PbF2, a parent of the broad class of ‘type II’ superionics. [2]. [1] H.J. Stroud, C.E. Mohn, J-A. Hernandez and N.L. Allan, Phil. Trans. Roy. Soc. A, 2021, 20200430
Professor Neil Allan, University of Bristol, UK
Professor Neil Allan, University of Bristol, UKNeil Allan has been a Professor of Physical Chemistry at the University of Bristol since 2003. He is a former Director of the Centre of Computational Chemistry (2008–2011). He has just completed five years as Chair of CCP5, the national Collaborative Computational Project for computer simulation of condensed phases. He was educated at Oxford University and then spent four years in industry working at ICI, before moving to Bristol in 1989. His main interests lie in the simulation materials of technological, geological and industrial interest, using a wide variety of theoretical techniques, ranging from atomistic lattice simulations to ab initio electronic structure calculations. Much of his work deals with the computed properties of inorganic materials and minerals, including solid solutions, the effects of additives, dopants and impurities, thin films and crystal growth. |
| 10:15 - 10:30 | Discussion |
| 10:30 - 11:00 | Break |
| 11:00 - 11:30 |
String-like relaxation in superionic conductors
Departing from the traditional quasi-static defect framework, the author provides considerable evidence for string-like dynamical structures, which govern the superionic conduction processes in fluorites and pyrochlores. The author shows that lower temperatures encourage the growth of longer but slowly relaxing strings and vice-versa; a direct manifestation of heterogeneous dynamics. The ionic conductivity is shown to be inversely correlated to the lifetime of the ions that participate in the strings. The current analysis methodology, which resolves a long-standing disagreement on defect structures and the mechanism of ionic transport in fcc fluorite structures, is well-positioned to describe the dynamics of low-dimensional conduction in a larger class of superionic conductors. 
Professor Jacob Eapen, North Carolina State University, USA
Professor Jacob Eapen, North Carolina State University, USADr Jacob Eapen joined the Department of Nuclear Engineering at the North Carolina State University in Spring 2008 following post-doctoral training at the Los Alamos National Laboratory. He obtained his Doctor of Science (ScD) degree in Nuclear Science and Engineering from the Massachusetts Institute of Technology in 2006. For a number of years, Dr Eapen has been working on thermal transport and glassy dynamics of supercooled liquids using atomistic simulations. More recently, he has been investigating the ‘thermally jammed’ nature of superionic conduction in fluorites, and phonon transport using atomistic simulations. He has been an investigator on several US Department of Energy (DOE) projects including accident tolerant fuel/clad with silicon carbide composites, and thermokinetics of molten salt mixtures. Dr Eapen has also served as a reviewer in several DOE programs. He currently serves as an executive member of the Materials Science and Technology Division of the American Nuclear Society. |
| 11:30 - 11:45 | Discussion |
| 11:45 - 12:15 |
Ionic motion in lithium thiophosphates studied by electronic structure calculations
Sulfide solid electrolytes (SE) are promising candidates for all-solid-state batteries because they show ionic conductivities of up to ≈10 mS/cm at room temperature and convince with favourable mechanical properties that enable an easy integration into the battery. Their disadvantage, however, is a lack of electrochemical stability against most electrode materials. Relevant processes, such as exact diffusion mechanisms or interface degradation reactions, have not been understood in detail. In this contribution, the author will present results on the lithium thiophosphate (LiPS) system that comprises multiple crystalline phases such as Li3PS4, Li7P3S11 and Li4P2S6, which coexist with amorphous phases in glass-ceramics. By means of electronic structure calculations within density functional theory the authors study the stability, structure and Li+ transport properties of crystalline and glassy phases. The authors' results reveal that all glasses are metastable and exhibit similar Li+ diffusion coefficients despite the fact that they are comprised of different basic structural units. The occurrence of unusual structural units is observed and the association of structural units via cross-linking S−S bonds is derived as compensation mechanism in case of local Li deficiency. Finally, the interfacial stability against Li metal and diffusion in grain boundaries are investigated. In this regard, the usage of defect formation energies as descriptors to judge the stability of interfaces is discussed. 
Professor Karsten Albe, Technische Universität Darmstadt, Germany
Professor Karsten Albe, Technische Universität Darmstadt, GermanyKarsten Albe has been a Professor of Materials Modelling at TU Darmstadt since 2002. He is a former Director of the Collaborative Research Centre 595 on Fatigue of Functional Materials (2012–2014) and is currently coordinating a research project on modelling of solid electrolytes funded by BMBF. He was educated in physics at the Universities of Hamburg and Ulm and was a PostDoc at the University of Illinois Urbana-Champaign after he finished is doctoral studies at TU Dresden. His main interests lie in the simulation defect of structures in functional materials using atomic scale models. Much of his work deals with ab-initio calculations of point defects, molecular dynamics simulations of disordered solids, dislocations and diffusional transport as well as the development of computational analysis tools. |
| 12:15 - 12:30 | Discussion |
| 13:30 - 14:00 |
Associate Professor Olivier Delaire, Duke University, USA
Abstract will be available soon. |
|---|---|
| 14:00 - 14:15 | Discussion |
| 14:15 - 14:45 |
Title of the talk will be available soon.
Ion diffusion is critical in a variety of important technologies, yet fundamental understanding of the interaction between the mobile species and dynamics of the host lattice is still missing. In this talk, the author will review some of the works on the roles of lattice dynamics on ionic conductivity starting from the early theories proposed by Mahan, Zeller, Rice and Roth in the 1970's and 1980's with the emphasis on various proposed correlations between Debye and Einstein frequency to more recent work on the role of specific vibrational modes on the activation energy. In particular, two formalisms to quantify the individual contributions of phonons to the ionic diffusion through a solid based on nudged elastic band calculations and molecular dynamics simulations will be presented. The application of these methods to a model ion conductor of Ge-substituted Li3PO4 in dilute limit reveal that more than 87% of the Li+ ion diffusion originates from less than 10% of the vibrational modes between 8 and 20 THz. By deliberately exciting a small, targeted subset of these contributing modes (<1%) to a higher temperature and keeping the lattice at a low temperature, we observe an increase in diffusivity by several orders of magnitude. This new understanding identifies new avenues to increase ion mobility by selectively excite vibrational modes having the strongest contribution to the diffusion in a material without necessarily changing the compound chemistry. Finally, several open questions regarding the fundamental understanding of the role of lattice dynamics and new strategies to tune ionic conductivity based on these concepts are presented. 
Dr Sokseiha Muy, EPFL, France
Dr Sokseiha Muy, EPFL, FranceSokseiha Muy received his bachelor's and master's degrees from Eìcole Polytechnique in France and a PhD in materials science and engineering from MIT under the supervision of Professor Yang Shao-Horn. He is currently a postdoctoral associate in the Theory and Simulation of Materials (THEOS) group with Professor Nicola Marzari at the Ecole Polytechnique Federale de Lausanne (EPFL), Switzerland. His research interests include the fundamental understanding of lattice dynamics on the ionic mobility in superionic conductors, design principle of new solid-state electrolytes for Li-ion battery, molecular dynamic simulations of ion conductors using machine-learning potentials and computational designs of defects in functional materials. |
| 14:45 - 15:00 | Discussion |
| 15:00 - 15:30 | Break |
| 15:30 - 16:00 |
Speaker to be confirmed soon.
|
| 16:00 - 16:15 | Discussion |
| 16:15 - 17:00 | Panel Discussion |